
Toda la función biológica depende de cómo las diferentes proteínas interactúen entre sí. Las interacciones proteína-proteína facilitan todo, desde la transcripción del ADN y el control de la división celular hasta las funciones de nivel superior en organismos complejos.
Sin secuestro, queda mucho sin estar claro sobre cómo estas funciones se orquestan en el nivel molecular y cómo interactúan las proteínas entre sí, ya sea con otras proteínas o con copias de sí mismas.
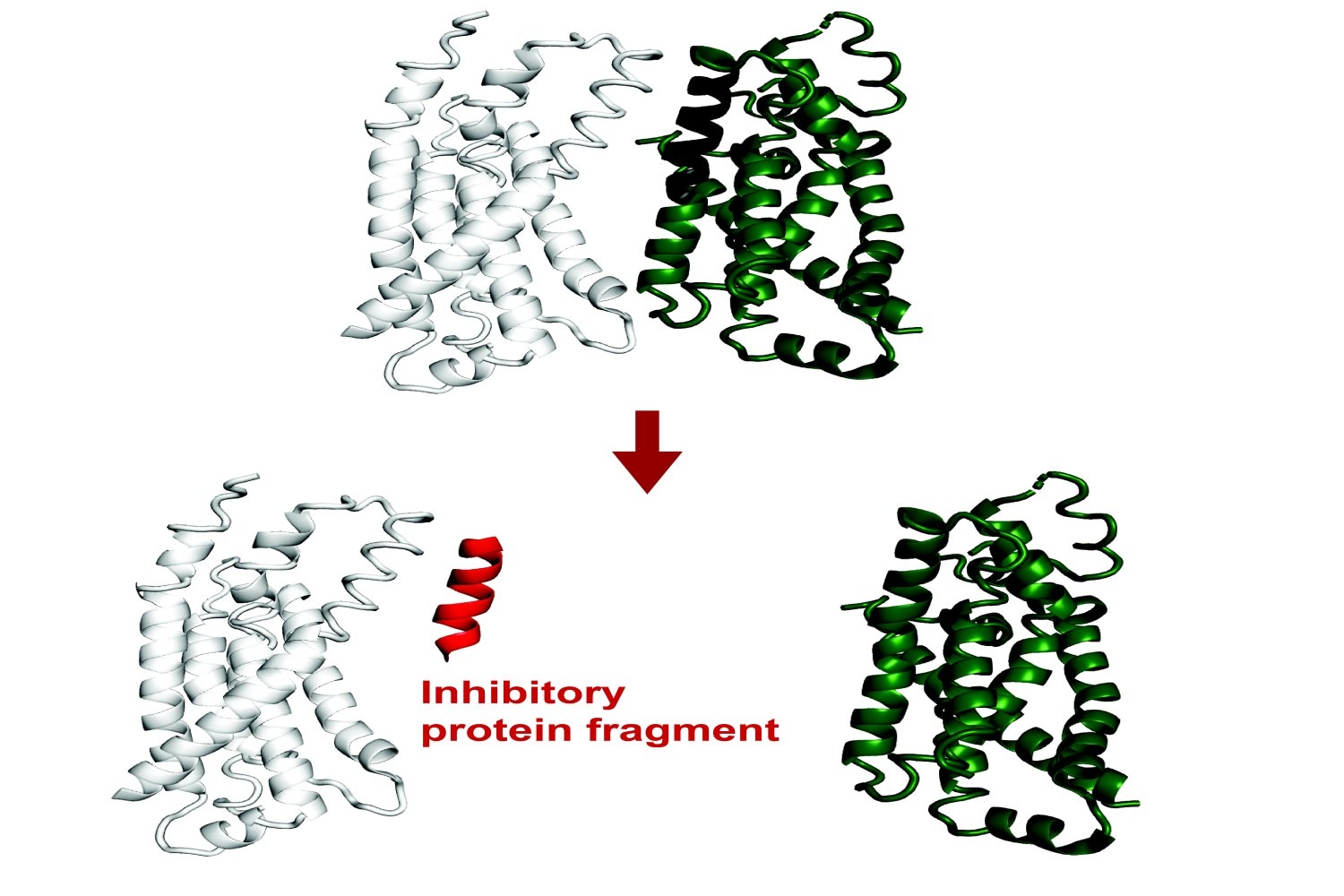
Hallazgos recientes han revelado que los fragmentos de proteínas pequeños tienen mucho potencial utilitario. A pesar de que son piezas incompletas, los tramos cortos de aminoácidos aún pueden unirse a las interfaces de una proteína objetivo, recapitulando las interacciones nativas. A través de este proceso, pueden alterar la función de esa proteína o interrumpir sus interacciones con otras proteínas.
Por lo tanto, los fragmentos de proteínas podrían empoderar tanto en la investigación básica sobre las interacciones de proteínas como los procesos celulares, y potencialmente podrían tener aplicaciones terapéuticas.
Recientemente publicado en Actas de la Agrupación Doméstico de Cienciasun nuevo método desarrollado en el Unidad de Biología se fundamento en modelos de inteligencia fabricado existentes para predecir computacionalmente fragmentos de proteínas que pueden unirse e inhibir las proteínas de largo completa en E. coli. Teóricamente, esta utensilio podría conducir a inhibidores genéticamente codificables contra cualquier proteína.
El trabajo se realizó en el laboratorio de profesor asociado de biología e investigador del Instituto Médico Howard Hughes Gene-Wei Li En colaboración con el laboratorio de Jay A. Stein (1968) Profesor de Biología, Profesor de Ingeniería Biológica y Director de Unidad Amy Keating.
Aprovechando el enseñanza espontáneo
El software, llamado Fragfold, aprovecha Alfafold, un maniquí de IA que ha llevado a avances fenomenales en biología en los últimos abriles oportuno a su capacidad para predecir el plegamiento de proteínas e interacciones proteicas.
El objetivo del tesina era predecir inhibidores de fragmentos, que es una aplicación novedosa de Alfafold. Los investigadores en este tesina confirmaron experimentalmente que más de la centro de las predicciones de Fragflet para la unión o la inhibición eran precisas, incluso cuando los investigadores no tenían datos estructurales previos sobre los mecanismos de esas interacciones.
«Nuestros resultados sugieren que este es un enfoque generalizable para encontrar modos de unión que probablemente inhiban la función de proteínas, incluso para nuevos objetivos de proteínas, y puede usar estas predicciones como punto de partida para más experimentos», dice el autor y el autor correspondiente. Andrew Savinov, un postdoc en el laboratorio LI. «Positivamente podemos aplicar esto a las proteínas sin funciones conocidas, sin interacciones conocidas, sin siquiera estructuras conocidas, y podemos poner poco de crédito en estos modelos que estamos desarrollando».
Un ejemplo es FTSZ, una proteína que es esencia para la división celular. Está proporcionadamente estudiado, pero contiene una región intrínsecamente desordenada y, por lo tanto, especialmente difícil de estudiar. Las proteínas desordenadas son dinámicas, y sus interacciones funcionales son muy fugaces, ocurriendo tan brevemente que las herramientas de biología estructural actuales no pueden capturar una sola estructura o interacción.
Los investigadores aprovecharon Fragfold para explorar la actividad de los fragmentos de FTSZ, incluidos los fragmentos de la región intrínsecamente desordenada, para identificar varias interacciones de unión nuevas con varias proteínas. Este brinco en la comprensión confirma y se expande en experimentos anteriores que miden la actividad biológica de FTSZ.
Este progreso es significativo en parte porque se realizó sin resolver la estructura de la región desordenada, y porque exhibe el poder potencial de fragdica.
«Este es un ejemplo de cómo Alfafold está cambiando fundamentalmente cómo podemos estudiar biología molecular y celular», dice Keating. «Las aplicaciones creativas de los métodos de IA, como nuestro trabajo en Fragfold, abren capacidades inesperadas y nuevas direcciones de investigación».
Inhibición y más allá
Los investigadores lograron estas predicciones fragmentando computacionalmente cada proteína y luego modelando cómo esos fragmentos se unirían a los socios de interacción que pensaban que eran relevantes.
Compararon los mapas de la unión predicha en toda la secuencia con los bienes de esos mismos fragmentos en las células vivas, determinadas usando mediciones experimentales de suspensión rendimiento en las que millones de células producen un tipo de fragmento de proteína.
Alphafold utiliza información coevolutiva para predecir el plegamiento, y típicamente evalúa la historia evolutiva de proteínas usando poco llamado alineaciones de secuencias múltiples para cada ejecución de predicción. Los MSA son críticos, pero son un cuello de botella para predicciones a gran escalera: pueden tomar una cantidad de tiempo y potencia computacional prohibitiva.
Para Fragfold, los investigadores en su oportunidad precalcularon el MSA para una proteína de largo completa una vez, y usaron ese resultado para llevar las predicciones para cada fragmento de esa proteína de largo completa.
Savinov, contiguo con el discente de Keating Lab Sebastian Swanson PhD ’23, predijo fragmentos inhibitorios de un conjunto diverso de proteínas encima de FTSZ. Entre las interacciones que exploraron se encontraba un confuso entre las proteínas de transporte de lipopolisacáridos LPTF y LPTG. Un fragmento de proteína de LPTG inhibió esta interacción, presumiblemente interrumpiendo el suministro de lipopolisacárido, que es un componente crucial del E. coli Membrana de células externas esencial para la aptitud celular.
«La gran sorpresa fue que podemos predecir la unión con tan suscripción precisión y, de hecho, a menudo predecir la unión que corresponde a la inhibición», dice Savinov. «Por cada proteína que hemos conocido, hemos podido encontrar inhibidores».
Los investigadores inicialmente se centraron en los fragmentos de proteínas como inhibidores porque si un fragmento podría cercar una función esencial en las células es un resultado relativamente simple para evaluar sistemáticamente. Mirando con destino a el futuro, Savinov asimismo está interesado en explorar la función de fragmentos fuera de la inhibición, como fragmentos que pueden estabilizar la proteína a la que se unen, mejoran o alteran su función, o desencadenan la degradación de la proteína.
Diseño, en principio
Esta investigación es un punto de partida para desarrollar una comprensión sistémica de los principios de diseño celular, y en qué utensilios pueden estar recurriendo modelos de enseñanza profundo para hacer predicciones precisas.
«Hay un objetivo más amplio y de maduro ámbito al que estamos construyendo», dice Savinov. «Ahora que podemos predecirlos, ¿podemos usar los datos que tenemos de predicciones y experimentos para extraer las características sobresalientes para descubrir qué ha aprendido Alfafold sobre lo que hace que un buen inhibidor?»
Savinov y los colaboradores asimismo profundizaron en cómo se unen los fragmentos de proteínas, explorando otras interacciones proteicas y mutando residuos específicos para ver cómo esas interacciones cambian cómo interactúa el fragmento con su objetivo.
Al examinar experimentalmente el comportamiento de miles de fragmentos mutados interiormente de las células, un enfoque conocido como escaneo mutacional profundo, reveló aminoácidos esencia responsables de la inhibición. En algunos casos, los fragmentos mutados fueron inhibidores aún más potentes que sus secuencias naturales de largo completa.
«A diferencia de los métodos anteriores, no nos limitamos a la identificación de fragmentos en datos estructurales experimentales», dice Swanson. “La fuerza central de este trabajo es la interacción entre los datos de inhibición real de suspensión rendimiento y los modelos estructurales predichos: los datos experimentales nos guían con destino a los fragmentos que son particularmente interesantes, mientras que los modelos estructurales predichos por Fragfold proporcionan una hipótesis específica y probable para cómo funcionan los fragmentos a nivel molecular «.
Savinov está entusiasmado con el futuro de este enfoque y sus innumerables aplicaciones.
«Al crear aglutinantes compactos y genéticamente codificables, Fragfold abre una amplia escala de posibilidades para manipular la función de la proteína», acuerda Li. «Podemos imaginar la entrega de fragmentos funcionalizados que pueden modificar proteínas nativas, cambiar su sede subcelular e incluso reprogramarlos para crear nuevas herramientas para estudiar biología celular y tratar enfermedades».